
Einführung in das Krankheitsbild
Morbus Pompe ist eine seltene, häufig mit schweren Beeinträchtigungen einhergehende Erbkrankheit, die sowohl im Kindes- als auch im Jugend- oder Erwachsenenalter auftreten kann. Bis jetzt sind je nach Lebensalter 3 verschiedene Formen bekannt:
- Infantiler Morbus Pompe
- Juveniler Morbus Pompe
- Adulter Morbus Pompe
Bei Patienten mit M. Pompe kann aufgrund eines genetischen (angeborenen) Defektes das Enzym α -1,4-Glukosidase (saure Maltase) nicht in ausreichender Menge gebildet werden. Das Enzym saure α -1,4-Glukosidase ist eines von mehreren Enzymen, die in den Lysosomen (kleine sackförmige Zellbestandteile) innerhalb der Zellen gebildet werden. Aufgabe von Enzymen ist es, bestimmte körpereigene Substanzen abzubauen, die entweder von Zellen weiterverwertet oder aus dem Körper ausgeschieden werden müssen. Ist nicht genügend Enzym vorhanden, um eine bestimmte Stärkeverbindung, das Glykogen, abzubauen, sammelt sich lysosomales Glykogen in zahlreichen Zelltypen und Geweben an und führt schließlich zu Fehlfunktionen in Organen.
Morbus Pompe ist die schwerste Form der bekannten 12 Glykogen-Speichererkrankungen und wird auch als Glykogenspeicherkrankheit Typ II, Glykogenose Typ II und "Myopathie bei Mangel an saurer Maltase" bezeichnet.
Wie bei über 40 weitere Muskelerkrankungen (sog. Muskeldystrophien) führt die Pompe-Krankheit zu Muskelschwund, daher liegt aufgrund des ursächlichen Defektes sowohl eine metabolische Myopathie (stoffwechselbedingte Muskelerkrankung) als auch eine lysosomale Speicherkrankheit vor.
Morbus Pompe ist zwar angeboren, der Zeitpunkt an dem die Krankheit ausbricht, und die Ausprägung der Symptomatik können aber von Fall zu Fall erheblich unterschiedlich sein. Bei der schwersten Verlaufsform „infantiler M. Pompe“ setzen die Symptome bereits in den ersten Lebensmonaten ein. Im Vordergrund steht eine massive Herzmuskelschwäche und aufgrund des deutlich reduzierten Muskeltonus wirken die Kinder schlaff (im englischen Sprachraum als "floppy baby" bezeichnet). Unbehandelt versterben viele bereits im ersten Lebensjahr an Herzversagen.
Tritt die Pompe-Erkrankung im Kindes-, Jugend-, oder Erwachsenenalter (mitunter erst relativ spät) in Erscheinung, ist das Beschwerdebild oft uneinheitlich und deutet zunächst aufgrund des Muskelschwundes auf das Vorliegen anderer neuromuskulärer Erkrankungen hin. Neben fortschreitender Muskelschwäche, kommt es in den meisten Fällen auch zu Atemproblemen aufgrund der Schwächung der Atemmuskulatur (Zwerchfell).
Während bei einigen Patienten nur leichte Krankheitszeichen mit geringfügigen Beeinträchtigungen auftreten, brauchen andere Patienten im späteren Verlauf Rollstühle und/oder Beatmung. Aufgrund der fortschreitenden Atemschwäche bis hin zum Atemversagen ist die Lebenserwartung bei Pompe-Patienten in den meisten Fällen verkürzt.
Schätzungen zufolge tritt die Erkrankung weltweit bei ungefähr 1 von 40.000 Lebendgeburten auf. Damit zählt M. Pompe zu den seltenen Krankheiten und ist auch in der Fachwelt unter der englischen Bezeichnung "Orphan Disease" (wörtlich: Waisenkinder der Medizin) bekannt.

Geschichte
Die Pompe-Krankheit wurde nach dem niederländischen Arzt und Pathologen J. C. Pompe (1901-1945) benannt. Er hat die Erkrankung 1932 bei einem 7 Monate alten Jungen erstmals als “Cardiomegalia glycogenica diffusa” beschrieben, ohne die weitreichenden Organbeteiligungen zu erkennen.
Symptomatik
Aufgrund des Mangels an der α-1,4-Glukosidase sammelt sich Glykogen in verschiedenen Organen an und führt zu entsprechenden Symptomen und zum charakteristischen klinischen Erscheinungsbild:
Muskelschädigungen:
Die Schädigung der Muskelzellen durch abgelagerte Speichersubstanz tritt meist schon kurz nach der Geburt in Erscheinung. Säuglinge fallen durch einen schlaffen Muskeltonus, die Unfähigkeit den Kopf selbständig anzuheben oder durch fehlende Krabbelaktivität auf. Bei späterem Einsetzen der ersten Symptome ist besonders die Muskulatur des Schulter- und Beckengürtels betroffen, was das Treppensteigen und das Heben der Arme über Schulterhöhe beeinträchtigt. Diese Symptome verschlechtern sich im Verlauf der Erkrankung durch die zunehmende Anreicherung von Glykogen in den Muskeln. Auch eine Vergrößerung der Zunge (Makroglossie) ist bei einigen Patienten zu beobachten. Da nicht nur die Muskeln, sondern auch die Nerven, die die Muskeln versorgen, geschädigt werden, kommt es zur Verminderung bis hin zum völligen Fehlen von Muskelreflexen (Areflexie).

Bei Säuglingen mit der infantilen Form des M. Pompe wird der Kopf beim Hochziehen an den Armen häufig nicht mitbewegt ("head lag").
Schluckbeschwerden:
Da auch die Muskulatur im Mund-, Rachen- und Halsbereich sowie die versorgenden Nerven geschädigt sein können, ist eine Beeinträchtigung der Schluckfähigkeit häufig. Oft fällt dieses Symptom bereits früh durch Probleme beim Saugen oder Füttern auf, später dann durch häufiges Verschlucken.
Atmungsstörungen:
Das Zwerchfell, der für den Atemvorgang wichtigste Muskel, kann je nach Schwere der Pompe-Erkrankung in seiner Funktion eingeschränkt sein. Dies beeinträchtigt die Atmung der Patienten, obwohl ihre Lungen völlig gesund sind. Durch die Zwerchfellschwäche kommt es zu abgeflachten Atemzügen und zu einer unzureichenden Belüftung der Lungen, was in der Folge eine höhere Infektionsanfälligkeit zur Folge hat. Bei vielen Patienten kommt es zu Luftnot (Dyspnoe) oder sogar zum Aussetzen der Atmung während der Nacht (Schlafapnoe).
Skelettbeschwerden:
Bei Pompe-Patienten trägt die Wirbelsäule eine größere Last, da sie nicht durch die Muskulatur unterstützt wird. Rückenschmerzen im unteren Wirbelsäulenbereich, Wirbelsäulenverkrümmungen oder Skoliosen sind die Folge.
Herzmuskelvergrößerung:
Bei Pompe- Patienten kann der Herzmuskel bereits früh geschädigt (Kardiomyopathie) und massiv vergrößert (Kardiomegalie) sein.
Lebervergrößerung:
Ein Teil der Pompe-Patienten zeigt auch eine Vergrößerung der Leber, da Glykogen auch in diesem Organ gespeichert sein kann.
Vererbung
M. Pompe wird autosomal rezessiv vererbt, das bedeutet, dass die Erkrankung nur zum Ausbruch kommt, wenn gleichzeitig zwei veränderte Gene – von jedem Elternteil eins – an die Nachkommen weitergegeben werden. Die Eltern selbst sind in den meisten Fällen gesund, nur sogenannte „Überträger“. Sie besitzen jeweils ein verändertes und ein unverändertes Gen.
Folgende Fälle sind möglich:
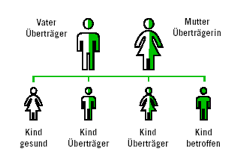
Wenn beide Eltern Träger eines fehlerhaften Gens (und damit Überträger der Erkrankung) sind, dann besteht bei jeder Schwangerschaft eine 25%ige Wahrscheinlichkeit, dass das Kind erkrankt. Mit ebenfalls 25%iger Wahrscheinlichkeit werden an das Kind zwei normale Gene weitergegeben. Dieses Kind ist dann völlig gesund, also auch kein Überträger der Erkrankung. In 50% der Fälle erbt ein Kind nur ein fehlerhaftes Gen. Dieses Kind ist dann zwar Überträger der Erkrankung, aber selbst nicht betroffen, da aufgrund des einen gesunden Gens genug Enzym gebildet werden kann, um die Speichersubstanz abzubauen.

Ist ein Elternteil gesund und ein Elternteil Überträger des fehlerhaften Gens, dann sind 50% der Kinder gesund, 50% der Kinder erbt jeweils ein gesundes und ein verändertes Gen und wird damit zum Überträger des veränderten Gens.
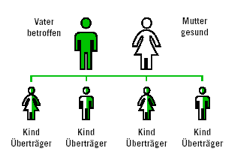
Wenn ein Elternteil an M. Pompe erkrankt und ein Elternteil gesund ist, dann erben alle Kinder je ein gesundes und ein verändertes Gen und werden zu Überträgern der Erkrankung.

Ist ein Elternteil Überträger und ein Elternteil an M. Pompe erkrankt, dann sind 50% der Kinder krank, 50% erben nur ein fehlerhaftes Gen und sind damit Überträger der Erkrankung, aber selbst nicht betroffen.